Ренин-ангиотензин-альдостероновая система (РААС) отвечает за норму объема экстрацеллюлярной жидкости, участвует в формировании стенок сосудов и обеспечивает уровень перфузии тканей. РААС непосредственно влияет на сердечно-сосудистую систему, нормализует артериальное давление и поддерживает содержание натрия и калия в норме.
В процессе участвует ренин (энзим), альдостерон (стероидный гормон) и ангиотензин II (пептидный гормон). Схема Ренин-ангиотензин-альдостероновой системы (РААС), представленная ниже, поможет понять принцип функционирования.
Основные цели РААС
Основной задачей для активации Ренин-ангиотензин-альдостероновой системы (РААС) является:
- Обеспечение достаточного кровотока в сосудах путем поддержания артериального давления, для функционирования печени, сердечно-сосудистой системы и сердца, почек, головного мозга.
- Выступает в роли «скорой помощи» при потере крови, при инфаркте и при резком снижении давления.
- Регулирует почечный и сосудистый гомеостаз, развивает процессы компенсаторного характера.
Длительная активация ренин-ангиотензин-альдостероновой системы может вызвать патологические явления в виде общего периферического сопротивления сосудов, недостаточный вывод жидкости из организма, избыток вырабатываемой крови, образованию периваскулярного и миокардиального фиброза.

Компонент системы ренин
Первым в звеньевой цепочке ренин-ангиотензин-альдостероновой системы, находится ренин, его производный элемент проренин, получается путем биосинтеза препрорениновой и рениновой РНК в юкстагломерулярных клетках. В дальнейшей подвергается глюкозелированию с последующим отщеплением аминокислот.
После деления, часть проренина выбрасывается в кровоток по принципу экзоцитоза, остаточная превращается в ренин, секретируясь юкстагломерулярными клетками аппарата почки, путем эндопептидаза. Образованный ренин в гранулах секреции юкстагломерулярной клетки в дальнейшем, также попадает в кровоток. Уровень производства ренина и дальнейшего поступления в кровь контролируется:
- артериальным давлением;
- химическими элементами NaClи Anq2;
- концентрацией внутри клетки ионов калия.

Ренин-ангиотензин-альдостероновая система призвана реагировать на сокращение объема воды и наличия натрия в организме при кровотечении. Потеря крови снижает давление в артериолах гломерулярных клубочков почек. Клетки стенок артериол улавливают спад натяжения, выделяют в капиллярную кровь ренин.
Большая часть регуляторов выработки ренина работает через почечные барорецепторы, под действием показателя состояния центральной нервной системы. На количество ренина влияет положение тела, переход из горизонтального положения в вертикальное или положение сидя, выработка энзима увеличивается. Это объясняется тем, что в симпатической части ЦНС повышается тонус и рефлекторно передается сигнал юкстагломерулярным клеткам.
В крови ренин воздействуя на ангиотензиноген, выделяет из него декапептид ангиотензин I, этот гормон не выполняет значимой функции в организме, но служит фундаментом для образования ангиотезина II. В процессе биохимической реакции, ангиотензин I, при помощи расщепления ангиотензинпревращающим ферментом (АПФ) переходит в ангиотезин II.
Ангиотезин II является центральным звеном ренин-ангиотензин-альдостероновой системы, основной задачей служит вазоконстрикторное воздействие на стенки артерий и ограниченное действие на центральную нервную систему. К рецепторам, участвующих в образование ангиотезина II, относятся следующие подтипы.
Агиотезин I-R (АТ 1-R) основа производного процесса, дает толчок основному количеству функций для реализации физиологически установленных норм ангиотезина II. Таким образом стимулируется выработка альдостерона надпочечниками, производится действие на симпатическую нервную систему. АТ 1-R мобилизирует ангиотезин II на рост клеток, и реагирование на воспалительный процесс. Влияние на сердечно-сосудистую систему, проявляется:
- повышением артериального давления;
- увеличением частоты сокращения сердечной мышцы;
- наличием сердечной и сосудистой гипертонии.

Следующий рецепторный тип АТ2-R к ангиотензину II, проявляет активность на первых стадиях развития эмбриона, при формировании мозга. На дальнейших этапах роста плода количество рецептора значительно сокращается.
Производный ангиотезина II – ангиотензиноген синтезируется печенью и под действием ренина, делится на ангиотезин I не активный декапептид, и на активный ангиотезин II, путем ферментативного воздействия АПФ. Функция активного октапептида ангиотезина II:
- путем сужения артериол повышает артериальное давление;
- контролирует выработку ренина юкстагломерулярными клетками;
- увеличивает сокращение миокарда;
- контролирует содержание натрия, ослаблением фильтрации в почках;
- поддержание водного баланса, путем формирования питьевого поведения.
Одной из важных задач ангиотезина II, воздействие на рецепторы центральной нервной системы, для активации биосинтеза в надпочечниках по выработке альдостерона. И путем обратной связи, всасывание ионов натрия почками.

Альдостерон
Синтез основного минералокортикоида происходит в гломерулярной зоне надпочечников, под воздействием калия и ангиотезина II и действует на мембранные рецепторы клеток ткани различных органов. Хотя основным производным альдостерона является ангиотезин II, сам гормон не участвует в производстве кортизола.
Функции альдостерона направлены на сдерживание натрия в почках и выведение лишнего количества натрия и калия из них. А также альдестерон играет немаловажную роль в ренин-ангиотензин-альдостероновой системе (РААС) отвечая:
- за защиту организма в неординарных ситуациях;
- стабилизирует уровень сахара в крови;
- сужение стенок сосуда, что делает невозможным понижение артериального давления, путем стабилизации кровяного потока.
Помимо регуляции артериального давления, альдостерон контролирует норму водно-солевого баланса. Но напрямую действуя на стенки сосудов, может вызвать нарушение функции эндотелия. Альдостерон способен спровоцировать воспаления стенки сосудов, активизировать моноциты крови и вызвать нарушение в почках и миокарде.
При повышенной выработке альдостерона или недостаточном количестве гормона необходимо медикаментозное лечение.
Для цитирования:
Леонова М.В. Новые и перспективные лекарственные препараты, блокирующие ренин-ангиотензин-альдостероновую систему // РМЖ. Медицинское обозрение. 2013. №17. С. 886
Роль ренин-ангиотензин-альдостероновой системы (РААС) в развитии артериальной гипертонии (АГ) и других сердечно-сосудистых заболеваний в настоящее время считается главенствующей. В кардиоваскулярном континууме АГ находится среди факторов риска, а главным патофизиологическим механизмом поражения сердечно-сосудистой системы является ангиотензин II (АТII). АТII является ключевым компонентом РААС - эффектором, который реализует вазоконстрикцию, задержку натрия, активацию симпатической нервной системы, клеточную пролиферацию и гипертрофию, развитие оксидативного стресса и процессов воспаления сосудистой стенки.
В настоящее время уже получили развитие и широкое клиническое применение два класса препаратов, блокирующих РААС, - ингибиторы АПФ и блокаторы рецепторов АТII. Фармакологические и клинические эффекты этих классов имеют отличия. АПФ является пептидазой из группы цинк-металлопротеиназ, которая метаболизирует АТI, АТ1-7, брадикинин, субстанцию Р и многие другие пептиды . Механизм действия ингибиторов АПФ главным образом связан с предотвращением образования АТII, что способствует вазодилатации, натрийурезу и устраняет провоспалительный, пролиферативный и другие эффекты АТII. Кроме того, ингибиторы АПФ препятствуют деградации брадикинина и повышают его уровень. Брадикинин - мощный вазодилататор, он потенцирует натрийурез, а главное - обладает кардиопротективным (предотвращает гипертрофию, уменьшает ишемическое повреждение миокарда, улучшает коронарное кровоснабжение) и вазопротективным действием, улучшая эндотелиальную функцию. Вместе с тем, высокий уровень брадикинина - причина развития ангионевротического отека, что является одним из серьезных недостатков ингибиторов АПФ, которые значительно повышают уровень кининов.
Ингибиторам АПФ не всегда удается полностью блокировать образование АТII в тканях. В настоящее время установлено, что в его превращении в тканях могут участвовать и другие ферменты, не связанные с АПФ, прежде всего эндопептидазы, на которые действие ингибиторов АПФ не распространяется. В результате ингибиторы АПФ не могут полностью устранить эффекты АТII, что может быть причиной их недостаточной эффективности.
Решению этой проблемы способствовало открытие рецепторов АТII и первого класса препаратов, селективно блокирующих АТ1-рецепторы. Через АТ1-рецепторы реализуются неблагоприятные эффекты АТII: вазоконстрикция, секреция альдостерона, вазопрессина, норадреналина, задержка жидкости, пролиферация гладкомышечных клеток и кардиомиоцитов, активация САС, а также механизм отрицательной «обратной связи» - образование ренина. АТ2-рецепторы выполняют «полезные» функции, такие как вазодилатация, процессы репарации и регенерации, антипролиферативное действие, дифференцировка и развитие эмбриональных тканей. Клинические эффекты блокаторов рецепторов АТII опосредованы через устранение «вредных» эффектов АТII на уровне АТ1-рецепторов, что обеспечивает более полное блокирование неблагоприятных эффектов АТII и усиление влияния АТII на АТ2-рецепторы, что дополняет вазодилатирующий и антипролиферативный эффекты. Блокаторы рецепторов АТII обладают специфичным действием на РААС, не вмешиваясь в кининовую систему. Отсутствие влияния на активность кининовой системы, с одной стороны, уменьшает выраженность нежелательных эффектов (кашель, ангионевротический отек), но, с другой, лишает блокаторы рецепторов АТII важного антиишемического и вазопротективного действия, что отличает их от ингибиторов АПФ. По этой причине показания к применению блокаторов рецепторов АТII в большинстве повторяют показания к назначению ингибиторов АПФ, делают их альтернативными препаратами.
Несмотря на внедрение блокаторов РААС в широкую практику лечения АГ, проблемы улучшения исходов и прогноза остаются. К ним относятся: возможность улучшения контроля АД в популяции, эффективность лечения резистентной АГ, возможности дальнейшего снижения риска сердечно-сосудистых заболеваний.
Поиск новых путей воздействия на РААС активно продолжается; изучаются другие тесно взаимодействующие системы и создаются препараты с множественным механизмом действия, такие как ингибиторы АПФ и нейтральной эндопептидазы (НЭП), ингибиторы эндотелин-превращающего фермента (ЭПФ) и НЭП, ингибиторы АПФ/НЭП/ЭПФ .
Ингибиторы вазопептидаз
К вазопептидазам кроме известного АПФ относятся еще 2 других цинк-металлопротеиназы - неприлизин (нейтральная эндопептидаза, НЭП) и эндотелин-превращающий фермент, которые также могут быть мишенями для фармакологического воздействия.
Неприлизин - фермент, вырабатываемый эндотелием сосудов и участвующий в деградации натрийуретического пептида, а также брадикинина.
Система натрийуретического пептида представлена тремя разными изоформами: предсердным натрий-уретическим пептидом (А-тип), мозговым натрийуретическим пептидом (В-тип), которые синтезируются в предсердии и миокарде, и эндотелиальным С-пептидом, которые по своим биологическим функциям являются эндогенными ингибиторами РААС и эндотелина-1 (табл. 1) . Кардиоваскулярные и ренальные эффекты натрийуретического пептида заключаются в снижении АД через влияние на сосудистый тонус и водноэлектролитный баланс, а также в антипролиферативном и антифибротическом действии на органы-мишени. По самым последним данным, система натрийуретического пептида участвует в метаболической регуляции: окислении липидов, образовании и дифференцировке адипоцитов, активации адипонектина, секреции инсулина и толерантности к углеводам, что может обеспечивать защиту от развития метаболического синдрома .
К настоящему времени стало известно, что развитие сердечно-сосудистых заболеваний ассоциируется с дизрегуляцией системы натрийуретического пептида. Так, при АГ наблюдается дефицит натрийуретического пептида, приводящий к солечувствительности и нарушению натрийуреза; при хронической сердечной недостаточности (ХСН) на фоне дефицита наблюдается аномалия функционирования гормонов системы натрийуретического пептида .
Поэтому для потенцирования системы натрийуретического пептида с целью достижения дополнительного гипотензивного и протективных кардиоренальных эффектов возможно применение ингибиторов НЭП. Ингибирование неприлизина приводит к потенцированию натрийуретического, диуретического и вазодилатирующего эффектов эндогенного натрийуретического пептида и в результате - к снижению АД. Однако НЭП участвует в деградации и других вазоактивных пептидов, в частности АТI, АТII и эндотелина-1. Поэтому баланс эффектов воздействия на сосудистый тонус ингибиторов НЭП вариабельный и зависит от преобладания констрикторных и дилатирующих влияний. При длительном применении антигипертензивное действие ингибиторов неприлизина выражено слабо вследствие компенсаторной активации образования АТII и эндотелина-1 .
В этой связи сочетание эффектов ингибиторов АПФ и ингибиторов НЭП может существенно потенцировать гемодинамические и антипролиферативные эффекты в результате комплементарного механизма действия, что привело к созданию препаратов с двойным механизмом действия, объединенных названием - ингибиторы вазопептидаз (табл. 2, рис. 1) .
Известные ингибиторы вазопептидаз характеризуются разной степенью селективности к НЭП/АПФ: омапатрилат - 8,9:0,5; фазидоприлат - 5,1:9,8; сампатрилат - 8,0:1,2 . В результате ингибиторы вазопептидаз получили гораздо большие возможности в достижении гипотензивного эффекта вне зависимости от активности РААС и уровня задержки натрия и в органопротекции (регресс гипертрофии, альбуминурии, жесткости сосудов). Наиболее изученным в клинических исследованиях был омапатрилат, который показал более высокую гипотензивную эффективность в сравнении с ингибиторами АПФ, а у пациентов с ХСН приводил к увеличению фракции выброса и улучшению клинических исходов (исследования IMPRESS, OVERTURE), но без преимуществ перед ингибиторами АПФ .
Однако в крупных клинических исследованиях с применением омапатрилата была установлена более высокая частота развития ангионевротического отека в сравнении с ингибиторами АПФ. Известно, что частота развития ангионевротического отека при использовании ингибиторов АПФ составляет от 0,1 до 0,5% в популяции, из них 20% случаев являются жизнеугрожающими, что связано с многократным повышением концентраций брадикинина и его метаболитов . Результаты крупного многоцентрового исследования OCTAVE (n=25 302), которое было специально спланировано для изучения частоты развития ангионевротического отека, показало, что частота развития этого побочного эффекта на фоне лечения омапатрилатом превышает таковую в группе эналаприла - 2,17% против 0,68% (относительный риск 3,4) . Это объяснялось усилением влияния на уровень кининов при синергичном ингибировании АПФ и НЭП, связанным с ингибированием аминопептидазы Р, участвующей в деградации брадикинина .
Новый двойной ингибитор вазопептидаз, блокирующий АПФ/НЭП, - илепатрил, который имеет более высокую аффинность к АПФ в сравнении с НЭП . При изучении фармакодинамических эффектов илепатрила по влиянию на активность РААС и натрийуретического пептида у здоровых добровольцев было установлено, что препарат дозозависимо (в дозах 5 и 25 мг) и значимо (более 88%) подавляет АПФ в плазме крови продолжительностью более 48 ч вне зависимости от солечувствительности. Одновременно препарат значимо повышал активность ренина плазмы в течение 48 ч и уменьшал уровень альдостерона . Эти результаты показали выраженное и более продолжительное подавление РААС в отличие от ингибитора АПФ рамиприла в дозе 10 мг, что объяснялось более значимым тканевым действием илепатрила на АПФ и большей аффинностью к АПФ, и сопоставимую степень блокады РААС в сравнении с комбинацией 150 мг ирбесартана + 10 мг рамиприла. В отличие от действия на РААС, эффект илепатрила на натрийуретический пептид проявлялся кратковременным увеличением уровня его экскреции в период 4-8 ч после приема дозы 25 мг, что свидетельствует о меньшей и слабой аффинности к НЭП и отличает его от омапатрилата. Причем по уровню экскреции электролитов дополнительного натрийуретического действия в сравнении с рамиприлом или ирбесартаном у препарата нет, как впрочем, и у других ингибиторов вазопептидаз. Максимальное гипотензивное действие развивается через 6-12 ч после приема препарата, и снижение среднего АД составляет 5±5 и 10±4 мм рт.ст. при низкой и высокой солечувствительности соответственно . По фармакокинетическим характеристикам илепатрил представляет собой пролекарство с активным метаболитом, который быстро образуется с достижением максимальной концентрации через 1-1,5 ч и медленно элиминирует. В настоящее время проводятся клинические исследования III фазы.
Альтернативный путь к двойному подавлению РААС и НЭП представлен сочетанием блокады рецепторов АТII и НЭП (рис. 2) . Блокаторы рецепторов АТII не влияют на метаболизм кининов в отличие от ингибиторов АПФ, поэтому потенциально имеют меньший риск развития ангионевротических осложнений. В настоящее время проходит фазу III клинических исследований первый препарат - блокатор рецепторов АТII с эффектом ингибирования НЭП в соотношении 1:1 - LCZ696. Объединенная молекула препарата содержит валсартан и ингибитор НЭП (AHU377) в форме пролекарства . В крупном исследовании у больных с АГ (n=1328) препарат LCZ696 в дозах 200-400 мг показал преимущество в гипотензивном эффекте перед валсартаном в дозах 160-320 мг в виде дополнительного снижения АД на 5/3 и 6/3 мм рт.ст. . Гипотензивный эффект LCZ696 сопровождался более выраженным снижением пульсового АД: на 2,25 и 3,32 мм рт.ст. соответственно в дозах 200 и 400 мг, что в настоящее время рассматривается как положительный прогностический фактор по влиянию на жесткость сосудистой стенки и сердечно-сосудистые исходы. При этом изучение нейрогуморальных биомаркеров на фоне лечения LCZ696 показало увеличение уровня натрийуретического пептида при сопоставимой степени увеличения уровня ренина и альдостерона в сравнении с валсартаном. Переносимость у больных с АГ была хорошей, и случаев ангионевротического отека не было отмечено. В настоящее время завершено исследование PARAMOUMT у 685 пациентов с ХСН и ненарушенной ФВ . Результаты исследования показали, что LCZ696 быстрее и выраженнее снижает уровень NT-proBNP (первичная конечная точка - маркер повышения активности натрий-уретического пептида и неблагоприятного прогноза при ХСН) в сравнении с валсартаном, а также уменьшает размеры левого предсердия, что свидетельствует о регрессе его ремоделирования . Исследование у пациентов с ХСН и сниженной ФВ продолжается в настоящее время (исследование PARADIGM-HF).
Ингибиторы системы эндотелина
Система эндотелина играет важную роль в регуляции сосудистого тонуса и регионального кровотока. Среди трех известных изоформ эндотелин-1 является наиболее активным. Кроме известных вазоконстрикторных эффектов эндотелин стимулирует пролиферацию и синтез межклеточного матрикса, а также вследствие прямого воздействия на тонус почечных сосудов участвует в регуляции водно-электролитного гомеостаза. Эффекты эндотелина реализуются через взаимодействие со специфическими рецепторами А-типа и В-типа, функции которых взаимопротивоположны: через А-тип рецепторов происходит вазоконстрикция, а через В-тип - вазодилатация . В последние годы установлено, что рецепторы В-типа играют большую роль в клиренсе эндотелина-1, т.е. при блокаде этих рецепторов нарушается рецепторзависимый клиренс эндотелина-1 и увеличивается его концентрация . Кроме того, рецепторы В-типа участвуют в регуляции почечных эффектов эндотелина-1 и поддержании водно-электролитного гомеостаза, что имеет важное значение.
В настоящее время роль эндотелина доказана в развитии ряда заболеваний, в т.ч. АГ, ХСН, легочной гипертензии, хронических заболеваний почек; показана тесная связь между уровнем эндотелина и метаболическим синдромом, дисфункцией эндотелия и атерогенезом. С 1990-х гг. ведется поиск антагонистов рецепторов эндотелина, пригодных для клинического использования; уже известно 10 препаратов («сентаны») с разной степенью селективности к А/B-типу рецепторов . Первый неселективный антагонист рецепторов эндотелина - бозентан - в клиническом исследовании у больных с АГ показал гипотензивную эффективность, сопоставимую с таковой ингибитора АПФ эналаприла . Дальнейшие исследования эффективности применения антагонистов эндотелина при АГ показали их клиническую значимость в лечении резистентной АГ и при высоком сердечно-сосудистом риске. Эти данные были получены в двух крупных клинических исследованиях DORADO (n=379) и DORADO-АС (n=849), в которых пациентам с резистентной АГ добавлялся дарусентан к тройной комбинированной терапии . В исследовании DORADO у пациентов резистентная АГ сочеталась с хронической болезнью почек и протеинурией, в результате добавления дарусентана наблюдалось не только значительное снижение АД, но и уменьшение экскреции белка. Антипротеинурический эффект антагонистов рецепторов эндотелина был в последующем подтвержден в исследовании у пациентов с диабетической нефропатией при использовании авосентана . Однако в исследовании DORADO-АС преимуществ в дополнительном снижении АД перед препаратами сравнения и плацебо не было выявлено, что послужило поводом к прекращению дальнейших исследований. Кроме того, в 4 крупных исследованиях антагонистов эндотелина (бозентана, дарусентана, энрасентана) у пациентов с ХСН были получены противоречивые результаты, что объяснялось увеличением концентрации эндотелина-1 . Дальнейшее изучение антагонистов рецепторов эндотелина было приостановлено ввиду нежелательных эффектов, связанных с задержкой жидкости (периферические отеки, перегрузка объемом). Развитие этих эффектов связывают с воздействием антагонистов эндотелина на В-тип рецепторов, что изменило поиск препаратов, влияющих на систему эндотелина через другие пути; а антагонисты рецепторов эндотелина в настоящее время имеют только одно показание - лечение легочной гипертензии.
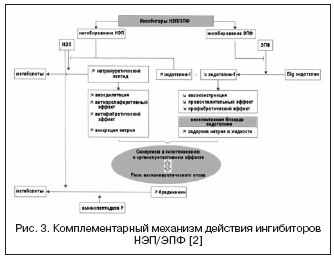
С учетом высокой значимости системы эндотелина в регуляции сосудистого тонуса ведется поиск другого механизма воздействия через вазопептидазу - ЭПФ, участвующий в образовании активного эндотелина-1 (рис. 3) . Блокирование ЭПФ и сочетание с ингибированием НЭП позволяют эффективно подавлять образование эндотелина-1 и потенцировать эффекты натрий-уретического пептида. Преимущества двойного механизма действия заключаются, с одной стороны, в предупреждении недостатков ингибиторов НЭП, связанных с возможной вазоконстрикцией, опосредованной активацией эндотелина, с другой, натрийуретическая активность ингибиторов НЭП позволяет компенсировать задержку жидкости, связанную с неселективной блокадой эндотелиновых рецепторов. Даглутрил является двойным ингибитором НЭП и ЭПФ, который находится во II фазе клинических исследований . В исследованиях показаны выраженные кардиопротективные эффекты препарата благодаря уменьшению ремоделирования сердца и сосудов, регрессу гипертрофии и фиброза.
Прямые ингибиторы ренина
Известно, что ингибиторы АПФ и блокаторы рецепторов АТII по механизму обратной связи повышают активность ренина, что является причиной ускользания эффективности блокаторов РААС. Ренин представляет собой самый первый этап каскада РААС; он вырабатывается юкстагломерулярными клетками почек. Ренин через ангиотензиноген способствует образованию АТII, вазоконстрикции и секреции альдостерона, а также регулирует механизмы обратной связи. Поэтому ингибирование ренина позволяет достичь более полной блокады системы РААС. Поиск ингибиторов ренина ведется с 1970-х гг.; долгое время не удавалось получить пер-оральную форму ингибиторов ренина ввиду их низкой биодоступности в ЖКТ (менее 2%). Первый прямой ингибитор ренина, пригодный для перорального применения, - алискирен - был зарегистрирован в 2007 г. Алискирен имеет низкую биодоступность (2,6%), большой период полувыведения (24-40 ч), внепочечный путь элиминации . Фармакодинамика алискирена связана с 80% уменьшением уровня АТII. В клинических исследованиях у пациентов с АГ алискирен в дозах 150-300 мг/сут приводил к снижению САД на 8,7-13 и 14,1-15,8 мм рт.ст. соответственно и ДАД - на 7,8-10,3 и 10,3-12,3 мм рт.ст. . Гипотензивный эффект алискирена наблюдался в разных подгруппах пациентов, включая больных с метаболическим синдромом, ожирением; по выраженности он был сопоставим с эффектом ингибиторов АПФ, блокаторов рецепторов АТII, а также отмечен аддитивный эффект в комбинации с валсартаном, гидрохлоротиазидом и амлодипином. В ряде клинических исследований были показаны органопротективные эффекты препарата: антипротеинурический эффект у пациентов с диабетической нефропатией (исследование AVOID, n=599) , регресс гипертрофии левого желудочка у пациентов с АГ (исследование ALLAY, n=465) . Так, в исследовании AVOID после 3-месячного лечения лозартаном в дозе 100 мг/сут и достижения целевого уровня АД (<130/80 мм рт.ст.) при компенсированном уровне гликемии (гликированный гемоглобин 8%) больных рандомизировали к приему алискирена в дозах 150-300 мг/сут или плацебо. Отмечено достоверное снижение индекса альбумин/креатинин в моче (первичная конечная точка) на 11% через 3 мес. и на 20% - через 6 мес. в сравнении с группой плацебо. В ночное время экскреция альбумина на фоне приема алискирена снизилась на 18%, а доля пациентов со снижением экскреции альбумина на 50% и более была вдвое большей (24,7% пациентов в группе алискирена против 12,5% в группе плацебо) . Причем нефропротективный эффект алискирена не был связан со снижением АД. Одним из объяснений выявленного нефропротективного эффекта у алискирена авторы считают полученные ранее в экспериментальных исследованиях на моделях диабета данные о способности препарата снижать количество рениновых и прорениновых рецепторов в почках, а также уменьшать профибротические процессы и апоптоз подоцитов, что обеспечивает более выраженный эффект в сравнении с эффектом ингибиторов АПФ . В исследовании ALLAY у пациентов с АГ и увеличением толщины миокарда ЛЖ (более 1,3 см по данным ЭхоКГ) применение алискирена ассоциировалось с одинаковой степенью регресса ИММЛЖ в сравнении с лозартаном и комбинацией алискирена с лозартаном: −5,7±10,6 , −5,4±10,8, −7,9±9,6 г/м2 соответственно. У части пациентов (n=136) проводилось изучение динамики нейрогормонов РААС, и было выявлено достоверное и значительное снижение уровня альдостерона и активности ренина плазмы на фоне применения алискирена или комбинации алискирена с лозартаном, тогда как на фоне применения монотерапии лозартаном эффект влияния на альдостерон отсутствовал, а на активность ренина - был противоположным, что объясняет значимость подавления альдостерона в достижении регресса ГЛЖ.
Кроме того, проводится серия клинических исследований алискирена при лечении других сердечно-сосудистых заболеваний с оценкой влияния на прогноз больных: исследования ALOFT (n=320), ASTRONAUT (n=1639), ATMOSPHERE (n=7000) у пациентов с ХСН, исследование ALTITUDE у пациентов с сахарным диабетом и высоким сердечно-сосудистым риском, исследование ASPIRE у пациентов с постинфарктным ремоделированием.
Заключение
Для решения проблем предупреждения сердечно-сосудистых заболеваний продолжается создание новых лекарственных препаратов со сложным множественным механизмом действия, позволяющих обеспечивать более полную блокаду РААС через каскад механизмов гемодинамической и нейрогуморальной регуляции. Потенциальные эффекты таких препаратов позволяют не только обеспечивать дополнительный гипотензивный эффект, но и достигать контроля уровня АД у пациентов высокого риска, включая резистентную форму АГ. Лекарственные препараты с множественным механизмом действия демонстрируют преимущества в более выраженном органопротективном действии, что позволит предупреждать дальнейшее поражение сердечно-сосудистой системы. Изучение преимуществ новых препаратов, блокирующих РААС, требует дальнейших исследований и оценки их влияния на прогноз больных с АГ и другими сердечно-сосудистыми заболеваниями.



Литература
1. Campbell D.J. Vasopeptidase inhibition: a doubleedged sword? // Hypertension. 2003. Vol. 41. P. 383-389.
2. Laurent S., Schlaich M., Esler M. New drugs, procedures, and devices for hypertension // Lancet. 2012. Vol. 380. P. 591-600.
3. Corti R., Burnett J.C., Rouleau J.L. et al. Vasopeptidase inhibitors: a new therapeutic concept in cardiovascular disease? // Circulation. 2001. Vol. 104. P. 1856-1862.
4. Mangiafico S., Costello-Boerrigter L.C., Andersen I.A. et al. Neutral endopeptidase inhibition and the natriuretic peptide system: an evolving strategy in cardiovascular therapeutics // Eur. Heart J. 2012, doi:10.1093/eurheartj/ehs262.
5. Rouleau J.L., Pfeffer M.A., Stewart D.J. et al. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial // Lancet. 2000. Vol. 356. P. 615-620.
6. Packer M., Califf R.M., Konstam M.A. et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: The Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE) // Circulation. 2002. Vol. 106. P. 920-926.
7. Warner K.K., Visconti J.A., Tschampel M.M. Angiotensin II receptor blockers in patients with ACE inhibitor-induced angioedema // Ann. Pharmacother. 2000. Vol. 34. P. 526-528.
8. Kostis J.B., Packer M., Black H.R. et al. Omapatrilat and enalapril in patients with hypertension:the Omapatrilat Cardiovascular Treatment vs Enalapril (OCTAVE) trial // Am. J. Hypertens. 2004. Vol. 17. P. 103-111.
9. Azizi M., Bissery A., Peyrard S. et al. Pharmacokinetics and pharmacodynamics of the vasopeptidase inhibitor AVE7688 in humans // Clin. Pharmacol. Ther. 2006. Vol. 79. P. 49-61.
10. Gu J., Noe A., Chandra P. et al. Pharmacokinetics and pharmacodynamics of LCZ696, a novel dualacting angiotensin receptorneprilysin inhibitor (ARNi) // J. Clin. Pharmacol. 2010. Vol. 50. P. 401-414.
11. Ruilope L.M., Dukat A., Buhm M. et al. Bloodpressure reduction with LCZ696, a novel dualacting inhibitor of the angiotensin II receptor and neprilysin: a randomised, double-blind, placebo-controlled, active comparator study // Lancet. 2010. Vol. 375. P. 1255-1266.
12. Solomon S.D., Zile M., Pieske B. et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial // Lancet. 2012. Vol. 380(9851). P. 1387-1395.
13. Levin E.R. Endothelins // N. Engl. J. Med. 1995. Vol. 333. P. 356-363.
14. Dhaun N., Goddard J., Kohan D.E. et al. Role of endothelin-1 in clinical hypertension: 20 years on // Hypertension. 2008. Vol. 52. P. 452-459.
15. Burnier M., Forni V. Endothelin receptor antagonists: a place in the management of essential hypertension? // Nephrol. Dial. Transplant. 2011. 0: 1-4. doi: 10.1093/ndt/gfr704.
16. Krum H., Viskoper R.J., Lacourciere Y. et al. The effect of an endothelin-receptor antagonist, bosentan, on blood pressure in patients with essential hypertension. Bosentan Hypertension Investigators // N. Engl. J. Med. 1998. Vol. 338. P. 784-790.
17. Weber M.A., Black H., Bakris G. et al. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: a randomised, double-blind, placebo-controlled trial // Lancet. 2009. Vol. 374. P. 1423-1431.
18. Bakris G.L., Lindholm L.H., Black H.R. et al. Divergent results using clinic and ambulatory blood pressures: report of a darusentan-resistant hypertension trial // Hypertension. 2010. Vol. 56. P. 824-830.
19. Mann J.F., Green D., Jamerson K. et al. Avosentan for overt diabetic nephropathy // J. Am. Soc. Nephrol. 2010. Vol. 21. P. 527-535.
20. Kalk P., Sharkovska Y., Kashina E. et al. Endothelinconverting enzyme/neutral endopeptidase inhibitor SLV338 prevents hypertensive cardiac remodeling in a blood pressure-independent manner // Hypertension. 2011. Vol. 57. P. 755-763.
21. Nussberger J., Wuerzner G., Jensen C. et al. Angiotensin II suppression in humans by theorally active renin inhibitor Aliskiren (SPP100): comparison with enalapril // Hypertension. 2002. Vol. 39(1). P. E1-8.
22. Alreja G., Joseph J. Renin and cardiovascular disease: Wornout path, or new direction? // World J. Cardiol. 2011. Vol. 3(3). P. 72-83.
23. Ingelfinger J.R. Aliskiren and dual therapy in type 2 diabetes mellitus // N. Engl. J. Med. 2008. Vol. 358(23). P. 2503-2505.
24. Pouleur A.С., Uno H., Prescott M.F., Desai A. (for the ALLAY Investigators). Suppression of aldosterone mediates regression of left ventricular hypertrophy in patients with hypertension // J. Renin-Angiotensin-Aldosterone System. 2011. Vol. 12. P. 483-490.
25. Kelly D.J., Zhang Y., Moe G. et al. Aliskiren, a novel renin inhibitor, is renoprotective in a model of advanced diabetic nephropathy in rats // Diabetol. 2007. Vol. 50. P. 2398-2404.
Ренин
– фермент, синтезируемый юкстагломерулярными клетками почечных афферентных артериол, имеющий ММ около 40 кДа. Особенно интенсивно образование ренина происходит при ишемии почек. Локализация юкстагломерулярных клеток делает их особенно чувствительными к изменениям кровяного давления, а также концентрации ионов Na + и К + в жидкости, протекающей через почечные канальцы. Благодаря указанным свойствам любая комбинация факторов, вызывающая снижение объема жидкости (обезвоживание, падение кровяного давления, кровопотеря и др.) или снижение концентрации NaCl, стимулирует высвобождение ренина.
В то же время большинство регуляторов синтеза ренина действуют через почечные барорецепторы. На освобождение ренина оказывает влияние состояние ЦНС, а также изменение положения тела в пространстве. В частности, при переходе из положения лёжа в положение сидя или стоя (клиностатическая проба) секреция ренина увеличивается. Эта рефлекторная реакция обусловлена повышением тонуса симпатической части автономной нервной системы, передающей импульсы к b-адренорецепторам юкстагломерулярных клеток.
Основным субстратом, на который воздействует ренин, является ангиотензиноген – белок, входящий во фракцию a 2 -глобулинов и образуемый печенью. Под воздействием глюкокортикоидов и эстрогенов синтез ангиотензиногена значительно возрастает. В результете действия ренина ангиотензиноген превращается в декапептид ангиотензин I. Это соединение обладает чрезвычайно слабым действием и существенного влияния на уровень кровяного давления не оказывает.
Между тем ангиотензин I под воздействием так называемого ангиотензинпревращающего фермента (АПФ) переходит в мощный сосудосуживающий фактор – ангиотензин II. АПФ (дипептидкарбооксипептидаза) является интегральным белком, расположенным преимущественно на мембране эндотелиальных клеток, эпителии, мононуклеарах, нервных окончаниях, клетках репродуктивных органов и др. Растворимая форма АПФ присутствует практически во всех жидкостях организма.
Принято выделять две изоформы АПФ. Первая из них получила условное наименование «соматической». Эта изоформа имеет ММ 170 кДа и включает гомологичные С- и N-домены. Вторая форма АПФ («репродуктивная») найдена в семенной жидкости, имеет ММ около 100 кДа и соответствует С-домену первой изоформы АПФ. Каждый из 2 указанных доменов содержит аминокислотные остатки, которые могут принимать участие в образовании связи с атомом цинка. Такие Zn 2+ -структуры являются типичными для многих металлопротеиназ и оказываются основными участками взаимодействия фермента как с субстратом, так и с ингибиторами АПФ.
Следует заметить, что АПФ не только приводит к образованию ангиотензина II, но и разрушает брадикинин – соединение, расширяющее кровеносные сосуды. Следовательно, увеличение кровяного давления при воздействии АПФ связано как с образованием ангиотензина II, так и с распадом брадикинина (рис. 32).
Важную роль для действия АПФ играет ионный состав и, в частности, содержание ионов хлора. Так, при высокой концентрации Cl – С-домен АПФ гидролизует и брадикинин, и ангиотензин-I быстрее, чем N-домен. Во внеклеточных регионах, где концентрация анионов хлора высока, за превращение ангиотензина-I отвечает преимущественно N-домен. Однако внутриклеточно, где концентрация Cl – низкая, N-домен может участвовать в гидролизе других пептидных субстанций.
За последние годы установлено, что АПФ играет важную роль в гемопоэзе, ибо под его воздействием ингибируется образование гематопоэтического пептида , тормозящего образование гематопоэтических клеток костного мозга.
Роль АПФ в организме была выявлена на мышах, лишенных гена АПФ. У таких животных отмечалось низкое кровяное давление, различные сосудистые дисфункции, нарушение структуры и функции почек и бесплодие у самцов.
Ангиотензин II
увеличивает кровяное давление, вызывая сужение артериол, и является самым сильнодействующим из известных вазоактивных агентов. Кроме того, он по механизму обратной связи тормозит образование и высвобождение ренина юкстагломерулярными клетками почки, что в конечном итоге должно восстанавливать нормальный уровень кровяного давления. Под воздействием ангиотензина II резко возрастает продукция основного минералокортикоида – альдостерона. Несмотря на то, что это действие является прямым, ангиотензин II не влияет на продукцию кортизола. Основное назначение альдостерона сводится к задержке Na + (за счет усиления его реабсорбции в почечных канальцах) и выделению К + и Н + (главным образом через почки). Эти реакции осуществляются следующим образом.
Альдостерон
проникает из внеклеточной жидкости в цитоплазму клетки и там соединяется со специфическим рецептором, после чего образовавшийся комплекс (альдостерон+рецептор) проникает в ядро. Альдостерон также стимулирует открытие Na + каналов, благодаря чему ионы Na + входят в клетку через апикальную мембрану из просвета канальца.
Увеличение секреции К + под воздействием альдостерона обусловлено возрастанием проницаемости апикальной мембраны по отношению к этим ионам, благодаря чему К + поступает из клетки в просвет канальца.
Задержка Na + в организме, как и ангиотензин II, способствует повышению кровяного давления.
Ангиотензин II способен связываться со специфическими рецепторами клубочковых клеток надпочечника. Содержание этих рецепторов во многом зависит от концентрации ионов К + . Так, если уровень К + повышается, то возрастает число рецепторов к ангиотензину II в клубочковых клетках. При уменьшении концентрации ионов К + отмечается прямо противоположный эффект. Следовательно, ионы К + играют основную роль в действии ангиотензина II на надпочечники.
За последнее время установлено, что ангиотензин II способен активировать макрофаги, благодаря чему усиливается агрегация тромбоцитов и ускоряется свёртывание крови. Одновременно при этом высвобождается ингибитор активатора плазминогена- I (ИАП-1), что может сопровождаться депрессией фибринолиза. Ангиотензин II является одним из факторов, способствующих развитию атерогенеза, торможению апоптоза и усилению оксидативного стресса в тканях, тем самым провоцируя агрегацию тромбоцитов и тромбообразование.
Ангиотензин II способен усиливать функцию миокарда, участвует в биосинтезе норадреналина и других физиологически активных веществ. Одновременно он может действовать как ростовой фактор, приводя к сосудистой и сердечной гипертрофии.
У некоторых животных и у человека ангиотензин II под воздействием фермента аминопептидазы превращается в гептапептид ангиотензин III. У человека уровень ангиотензина II приблизительно в 4 раза выше, чем ангиотензина III. Оба эти соединения оказывают влияние на уровень кровяного давления и продукцию альдостерона и довольно быстро разрушаются под воздействием ферментов ангиотензиназ.
При тяжелых заболеваниях почек, сопровождающихся их ишемией, благодаря повышенному образованию и секреции ренина наблюдается стойкое повышение кровяного давления (почечная гипертензия). Применение ингибиторов АПФ в этих условиях приводит к быстрой нормализации кровяного давления.
В заключение следует еще раз подчеркнуть, что ангиотензин-ренино-альдостероновая система теснейшим образом связана с функцией калликреин-кининовой системы, ибо образование ангиотензина II и разрушение брадикинина осуществляется под воздействием одного и того же фермента – АПФ.
Ренин-ангиотензин-альдостероновая система является комплексом ферментов и гормонов, которые поддерживают гомеостаз. Регулирует равновесие соли и воды в организме и уровень артериального давления.
Механизм работы
Физиология ренин-ангиотензин-альдостероновой системы берет начало на границе коркового и где имеются юкстагломерулярные клетки, вырабатывающие пептидазу (фермент) - ренин.
Ренин является гормоном и начальным звеном РААС.
Ситуации, при которых ренин выделяется в кровь
Существует несколько состояний, при которых идет попадание гормона в кровеносное русло:
- Уменьшение кровотока в ткани почек - при воспалительных процессах (гломерулонефрит др.), при диабетической нефропатии, опухолях почек.
- Снижение (при кровотечении, многократной рвоте, поносах, ожогах).
- Падение уровня артериального давления. В артериях почек имеются барорецепторы, которые реагируют на изменение системного давления.
- Изменение концентрации ионов натрия. В организме человека имеются скопления клеток, которые отвечают на изменение ионного состава крови стимуляцией выработки ренина. Соль теряется при обильном потоотделении, а также при рвоте.
- Стрессы, психоэмоциональные нагрузки. почки иннервируется симпатическими нервами, которые активируются при негативных психологических влияниях.
В крови ренин встречается с белком - ангиотензиногеном, который вырабатывается клетками печени и забирает у него фрагмент. Образуется ангиотензин I, который является источником воздействия для ангиотензинпревращающего фермента (АПФ). В итоге получается ангиотензин II, который служит вторым звеном и является мощным вазоконстриктором артериальной системы (суживает сосуды).
Эффекты ангиотензина II
Цель: повысить артериальное давление.
- Способствует синтезу альдостерона в клубочковой зоне коры надпочечников.
- Воздействует на центр голода и жажды в головном мозге, вызывая "солевой" аппетит. Поведение человека становится мотивированным на поиск воды и соленой пищи.
- Влияет на симпатические нервы, способствуя высвобождению норадреналина, который тоже является вазоконстриктором, но менее слабым по действию.
- Воздействует на сосуды, вызывая их спазм.
- Участвует в развитии хронической сердечной недостаточности: способствует пролиферации, фиброзу сосудов и миокарда.
- Снижает
- Тормозит выработку брадикинина.

Альдостерон - третий компонент, который действует на конечные канальцы почек и способствует выделению из организма ионов калия, магния и обратному всасыванию (реабсорбции) натрия, хлора, воды. Благодаря этому возрастает объем циркулирующей жидкости, поднимаются цифры артериального давления, и усиливается почечный кровоток. Рецепторы к альдостерону имеются не только в почках, но и в сердце, сосудах.
Когда организм достигает гомеостаза, начинают вырабатываться вазодилататоры (вещества, расширяющие сосуды) - брадикинин и каллидин. А компоненты РААС разрушаются в печени.
Схема ренин-ангиотензин-альдостероновой системы

Как любая система, РААС может давать сбой. Патофизиология ренин-ангиотензин-альдостероновой системы проявляет при следующих состояниях:
- Поражение коры надпочечников (инфекция, кровоизлияние и травма). Развивается состояние нехватки альдостерона, и организм начинает терять натрий, хлор и воду, что приводит к уменьшению объема циркулирующей жидкости и снижению артериального давления. Состояние компенсируют введением солевых растворов и стимуляторов рецепторов к альдостерону.
- Опухоль коры надпочечников приводит к избытку альдостерона, который реализует свои эффекты и повышает давление. Также активизируются процессы деления клеток, возникает гипертрофия и фиброз миокарда, и развивается сердечная недостаточность.
- Патология печени, когда нарушается разрушение альдостерона и происходит его накопление. Патология лечится блокаторами рецепторов к альдостерону.
- Воспалительные заболевания почек.

Значение РААС для жизни и медицины
Ренин-ангиотензин-альдостероновая система и ее роль в организме:
- принимает активное участие в поддержании нормального показателя артериального давления;
- обеспечивает равновесие воды и солей в организме;
- поддерживает кислотно-основной баланс крови.
Система может давать сбой. Воздействуя на ее компоненты, можно бороться с гипертонической болезнью. Механизм возникновения почечной гипертензии также тесно связан с РААС.
Высокоэффективные группы препаратов, которые синтезированы благодаря изучению РААС
- "Прилы". АПФ. Ангиотензин I не переходит в ангиотензин II. Нет вазоконстрикции - нет повышения артериального давления. Препараты: Амприлан, Эналаприл, Каптоприл и др. Ингибиторы АПФ значительно улучшают качество жизни больных сахарным диабетом, обеспечивая профилактику почечной недостаточности. Препараты принимают в минимальной дозировке, которая не вызывает снижения давления, а лишь улучшает местный кровоток и клубочковую фильтрацию. Медикаменты незаменимы при почечной недостаточности, хронической болезни сердца и служат одним из средств лечения гипертонической болезни (если нет противопоказаний).
- "Сартаны". Блокаторы рецепторов к ангиотензину II. Сосуды не реагируют на него и не сокращаются. Препараты: Лозартан, Эпросартан и др.

Противоположной ренин-ангиотензин-альдостероновой системе является кининовая. Поэтому блокирование РААС приводит к повышению в крови компонентов кининовой системы (брадикинин и др.), что благоприятно влияет на ткани сердца и стенки сосудов. Миокард не испытывает голодания, потому как брадикинин усиливает местный кровоток, стимулирует выработку естественных вазодилататоров в клетках мозгового вещества почек и микроцитах собирательных трубочек - простагландинов Е и И2. Они нейтрализуют прессорное действие ангиотензина II. Сосуды не спазмированы, что обеспечивает адекватное кровоснабжение органов и тканей организма, кровь не задерживается и снижается формирование атеросклеротических бляшек и тромбов. Кинины благоприятно воздействуют на почки, увеличивают диурез (суточное выделение мочи).
Ренин-ангиотензин-альдостероновая система (РААС.)
В регуляции объема и давления крови участвует юкстагломерулярный аппарат (ЮГА). Образующийся в гранулах клеток ЮГА протеолитический фермент ренин катализирует превращение ангиотензиногена (одного из белков плазмы) в декапептид ангиотензин I, который не обладает прессорной активностью. Под действием ангиотензин-превращающего фермента (АПФ) он расщепляется (главным образом в легких, почках, головном мозге) до октапептида ангиотензина II, который действует как мощный вазоконстриктор, а также стимулирует выработку альдостерона корой надпочечников. Альдостерон усиливает реабсорбцию Nа+ в канальцах почек и стимулирует выработку антидиуретического гормона. В результате чего происходит задержка Nа+ и воды, что приводит к повышению АД. Кроме того, в плазме крови имеется ангиотензин III (гептапептид, не содержащий аспарагиновой кислоты), который также активно стимулирует высвобождение альдостерона, но обладает менее выраженным прессорным действием, чем ангиотензин II. Следует отметить, что чем больше образуется ангиотензина II, тем сильнее выражена вазоконстрикция и, следовательно, тем более выражено повышение АД.
Секреция ренина регулируется следующими механизмами, не являющимися взаимоисключающими:
- 1) барорецепторами почечных сосудов, которые, очевидно, реагируют на изменение напряжения стенки приносящих артериол,
- 2) рецепторами macula densa, которые, по-видимому, чувствительны к изменению скорости поступления или концентрации NaCl в дистальных канальцах,
- 3) отрицательной обратной связью между концентрацией в крови ангиотензина и секрецией ренина
- 4) симпатической нервной системой, стимулирующей секрецию ренина в результате активации в-адренорецепторов почечного нерва.
Система поддержания гомеостаза натрия. Она включает в себя скорость клубочковой фильтрации (СКФ) и факторы натрийуреза (выведения ионов натрия с мочой). При снижении ОЦК, снижается и СКФ, что приводит, в свою очередь, к повышению реабсорбции натрия в проксмальном отделе нефрона. К факторам натрийуреза относится группа пептидов со схожими свойствами и общим названием - натрийуретический пептид (или атриопептид), вырабатываемых миокардом предсердий в ответ на их расширение. Эффект атриопептида заключается в уменьшении реабсорбции натрия в дистальных канальцах и вазодилятации.
Система почечных вазодепрессорных субстанций включает: простагландины, калликреин-кининовая система, NO, фактор активации тромбоцитов, которые своим действием уравновешивают вазопрессорный эффект ангиотензина.
Кроме того, определенную роль в манифестации АГ играют такие факторы внешней среды (рис.1 пункт 6), как гиподинамия, курение, хронические стрессы, избыточное потребление с пищей поваренной соли.
Этиология артериальной гипертензии:
Этиология первичной, или эссенциальной, гипертензии не известна. И вряд ли одна причина смогла бы объяснить такое разнообразие гемодинамических и патофизиологических расстройств, которые наблюдаются при данном заболевании. В настоящее время многие авторы придерживаются мозаичной теории развития АГ, согласно которой поддержание высокого АД обусловлено участием многих факторов, даже если первоначально доминировал какой-либо один из них (например, взаимодействие симпатической нервной системы и ренин-ангиотензин-альдостероновой системы).
Не вызывает сомнения, что существует генетическая предрасположенность к гипертензии, однако точный механизм ее до сих пор не ясен. Возможно, что факторы внешней среды (такие как количество натрия в пище, характер питания и образ жизни, способствующие ожирению, хронический стресс) оказывают свое действие только на генетически предрасположенных лиц.
Основные причины развития эссенциальной гипертензии (или гипертонической болезни) на долю которой приходится 85-90% случаев всех АГ следующие:
- - активация ренин-ангиотензин-альдостероновой системы при изменениях в генах, кодирующих ангиотензиноген или другие белки РААС,
- - активация симпатической нервной системы, что приводит к повышению АД преимущественно путем вазоконстрикции,
- - нарушение транспорта Na+ через клеточные мембраны гладкомышечных клеток кровеносных сосудов (в результате торможения Na+-K+-насоса или повышения проницаемости мембран для Na+ с повышением содержания внутриклеточного Са2+),
- - дефицит вазодилятаторов (таких, как NO, компоненты калликреин-кининовой системы, простагландины, предсердный натрийуретический фактор и др.).
Среди основных причин симптоматических гипертензий можно выделить:
- - первичное двустороннее поражение почек (которое может сопровождаться АГ вследствие как повышения секреции ренина и активации РААС с задержкой натрия и жидкости, так и снижения секреции вазодилятаторов) при таких заболеваниях, как острый и хронический гломерулонефрит, хронический пиелонефрит, поликистоз почек, амилоидоз, опухоли почек, обструктивная уропатия, коллагенозы и др.
- - эндокринные (потенциально излечимые) заболевания, такие как первичный и вторичный гиперальдостеронизм, болезнь и синдром Иценко-Кушинга, диффузный тиреотоксический зоб (Базедова болезнь или болезнь Грейвса), феохромоцитома, ренин-продуцирующие опухоли почек.
- - нейрогенные заболевания, в том числе сопровождающимися повышением внутричерепного давления (травма, опухоль, абсцесс, кровоизлияния), поражением гипоталамуса и ствола мозга, связанные с психогенными факторами.
- - сосудистые заболевания (васкулиты, коарктация аорты и другие аномалии сосудов), полицитемия, увеличение ОЦК ятрогенного характера (при избыточном переливании препаратов крови и растворов).
Морфология артериальной гипертензии:
Доброкачественная форма АГ:
На ранних стадиях АГ не удается обнаружить никаких структурных изменений. В конечном же итоге развивается генерализованный артериолярный склероз.
Учитывая длительное течение болезни, выделяют три стадии, имеющие определенные морфологические различия и согласующиеся со стадиями, предложенными экспертами ВОЗ (указанными в скобках):
- 1) доклиническая (легкое течение),
- 2) распространенных изменений артерий (средней тяжести),
- 3) изменений органов в связи изменением артерий и нарушением органного кровотока (тяжелое течение) доклиническая стадия.
Клинически проявляется транзиторной гипертензией (эпизодами повышения АД). На ранней, лабильной, стадии болезни СВ увеличен, ОПСС некоторое время остается в пределах нормы, но неадекватно для данного уровня СВ. Затем, вероятно в результате процессов ауторегуляции, ОПСС начинает увеличиваться, а СВ возвращается к нормальному уровню.
В артериолах и мелких артериях выявляется гипертрофия мышечного слоя и эластических структур > постепенное ^ толщины стенки сосуда с уменьшением его просвета, что клинически проявляется в ^ ОПСС. Спустя некоторое время на фоне катехолемии, ^ гематокрита, гипоксии (элементов стенки артерий и артериол) повышается сосудистая проницаемость, что приводит к плазматическому пропитыванию стенки сосудов > уменьшению ее эластичности и еще большему ^ ОПСС. Морфологические изменения на данной стадии полностью обратимы и при своевременном начале антигипертензивной терапии возможно предотвратить развитие поражений органов-мишеней.
В сердце, вследствие транзиторного ^ постнагрузки, возникает умеренная компенсаторная гипертрофия левого желудочка при которой размеры сердца и толщина стенки левого желудочка ^, а размер полости левого желудочка не изменяется либо может несколько уменьшаться - концентрическая гипертрофия (характеризует стадию компенсации сердечной деятельности).
Стадия распространенных изменений артерий. Клинически проявляется стойким повышением АД.
В артериолах и мелких артериях мышечного типа выявляется распространенный гиалиноз, развившийся в исходе плазматического пропитывания (простой тип сосудистого гиалина), или артериолосклероз средней оболочки и интимы артериол в ответ на выход плазмы и белков. Артериологиалиноз отмечается в почках, головном мозге, сетчатке глаза, поджелудочной железе, кишечнике, капсуле надпочечников. Макроскопически гиалинизированные сосуды выглядят в виде стекловидных трубочек с толстыми стенками и точечным просветом, плотной консистенции. Микроскопически в стенке артериол выявляются гомогенные эозинофильные массы, слои стенки могут быть практически не различимы.
В артериях эластического, мышечно-эластического и мышечного типов развиваются: - эластофиброз - гиперплазия и расщепление внутренней эластической мембраны, склероз - атеросклероз, имеющий ряд особенностей:
- а) носит более распространенный характер, захватывает артерии мышечного типа,
- б) фиброзные бляшки имеют циркулярный характер (а не сегментарный), что приводит к более значительному сужению просвета сосуда.
В сердце нарастает степень гипертрофии миокарда, масса сердца может достигать 900-1000 г, а толщина стенки левого желудочка - 2-3 см (cor bovinum). Однако, в связи с относительной недостаточностью кровоснабжения (увеличение размеров кардиомиоцитов, гиалиноз артериол и артерий) и нарастающей гипоксией развивается жировая дистрофия миокарда и миогенное расширение полостей - эксцентрическая гипертрофия миокарда, диффузный мелкоочаговый кардиосклероз, появляются признаки сердечной декомпенсации.
3) Стадия изменений органов в связи изменением артерий и нарушением органного кровотока.
Вторичные изменения органов при неосложенном артериологиалинозе и атеросклерозе могут развиваться медленно, что приводит к атрофии паренхимы и склерозу стромы.
При присоединении тромбоза, спазма, фибриноидного некроза во время криза возникают острые нарушения кровообращения - кровоизлияния, инфаркты.
Изменения в головном мозге:
Множественные мелкоочаговые кровоизлияния (hemorragia per diapedesin).
Гематомы - кровоизлияния с разрушением ткани мозга (hemorragia per rhexin микроанавризм, которые возникают чаще на фоне гиалиноза с фибриноидным некрозом стенки мелких перфорирующих артерий головного мозга преимущественно подкорковых ядер и субкортикального слоя). В исходе кровоизлияний в ткани головного мозга формируются ржавые кисты (окраска обусловлена гемосидерином).
В почках развивается артериолосклеротический нефросклероз или первичное сморщивание почек, в основе которого лежит артериологиалиноз >запустевание со склерозом и гиалинозом капилляров клубочков > склероз стромы вследствие длительной гипоксии > атрофия эпителия канальцев почек.
Макроскопическая картина: почки значительно уменьшены в размерах (вид местной атрофии от недостатка кровоснабжения), поверхность мелкозернистая, плотные, на разрезе отмечается истончение коркового и мозгового слоев, разрастание жировой клетчатки вокруг лоханки. Участки западения на поверхности почек соответствуют атрофированным нефронам, а очаги выбухания - функционирующим нефронам в состоянии компенсаторной гипертрофии.
Микроскопическая картина: стенки артериол значительно утолщены за счет накопления в интиме и средней оболочке гомогенных слабооксифильных бесструктурных масс гиалина (в некоторых случаях структурные компоненты стенки артериол, за исключением эндотелия, не дифференцируются), просвет сужен (вплоть до полной облитерации). Клубочки коллабированы (спавшиеся), многие замещены соединительной тканью или массами гиалина (в виде слабооксифильных гомогенных «медальончиков»). Канальцы атрофированы. Количество интерстициальной ткани увеличено. Сохранившиеся нефроны компенсаторно гипертрофированы.
Артериолосклеротический нефросклероз может закончиться развитием хронической почечной недостаточности.
Злокачественная форма АГ:
В настоящее время встречается редко.
Возникает первично или осложняет доброкачественную гипертензию (гипертонический криз).
Клинически: уровень Рдиаст.? 110-120 мм рт. ст., зрительные расстройства (из-за двустороннего отека диска зрительного нерва), резкие головные боли и гематурия (реже - анурия).
Уровень ренина и ангиотензина II в сыворотке крови высокий, значительный вторичный гиперальдстеронизм (сопровождающийся гипокалиемией).
Возникает чаще у мужчин среднего возраста (35-50 лет, редко до 30-ти лет).
Быстро прогрессирует, без лечения приводит к развитию хронической почечной недостаточности (ХПН) и летальному исходу в течение 1-2 лет.
Морфологическая картина:
Вслед за короткой стадией плазматического пропитывания следует фибриноидный некроз стенки артериол >повреждение эндотелия > присоединение тромбоза > органные изменения: ишемическая дистрофия и инфаркты, кровоизлияния.
Со стороны сетчатки: двусторонний отек диска зрительного нерва, сопровождающийся белковым выпотом и кровоизлияниями в сетчатку
В почках: злокачественный нефросклероз (Фара), для которого характерны фибриноидный некроз стенок артериол и капиллярных петель клубочков, отек интерстиция, геморрагии > клеточная реакция и склероз в артериолах, клубочках и строме, белковая дистрофия эпителия канальцев почек.
Макроскопическая картина: вид почек зависит от длительности предсуществующей фазы доброкачественной АГ. В связи с этим, поверхность может быть гладкой или гранулированной. Весьма характерны петехиальные кровоизлияния, которые придают почке пестрый вид. Прогрессирование дистрофических и некротических процессов быстро приводит к развитию ХПН и смерти.
В головном мозге: фибриноидный некроз стенок артериол с присоединением тромбоза и развитием ишемических и геморрагических инфарктов, кровоизлияний, отек.
Гипертонический криз - резкое повышение АД, связанное со спазмом артериол - может возникать в любой стадии гипертензии.
Морфологические изменения при гипертоническом кризе:
Спазм артериол: гофрированность и деструкция базальной мембраны эндотелия с расположением его в виде частокола.
Плазматическое пропитывание.
Фибриноидный некроз стенок артериол.
Диапедезные кровоизлияния.
Клинико-морфологические формы АГ:
В зависимости от преобладания сосудистых, дистрофических, некротических, геморрагических и склеротических процессов в том или ином органе, выделяют следующие формы:
Сердечная форма - составляет сущность ишемической болезни сердца (как и сердечная форма атеросклероза)
Мозговая форма - лежит в основе большинства цереброваскулярных заболеваний (как и атеросклероз сосудов головного мозга)
Почечная форма характеризуется как острыми (артериолонекроз - морфологическое проявление злокачественной гипертензии), так и хроническими изменениями (артериолосклеротический нефросклероз).
Рис. 1
Список сокращений к лекции «Гипертоническая болезнь»
АГ - артериальная гипертензия.
АД - артериальное давление.
ОЦК - объем циркулирующей крови.
СВ - сердечный выброс.
ОПСС - общее периферическое сопротивление сосудов.
УО - ударный объем.
ЧСС - частота сердечных сокращений.
СНС - симпатическая нервная система.
ПСНС - парасимпатическая нервная система.
РААС - ренин-ангиотензин-альдостероновая система.
ЮГА - юкстагломерулярный аппарат.
АПФ - ангиотензин-превращающий фермент.
СКФ - скорость клубочковой фильтрации.
ВОЗ - всемирная организация здравоохранения.
ХПН - хроническая почечная недостаточность.